Chemical Descriptors Library: applications
|
|
Miscellaneous applications
|
 |
nails .- cheminformatics tools
Contents
- Introduction
- Molecular Formats
- Descriptor Calculation
- Fingerprints
- Pharmacophore
- Maximum Overlapping Sets
- Structure Modification
- Synergies
- Support Vector Machines
- Neural Networks
- Challenges and TODO
- References
Nails is a Chemical Descriptors Library (CDL)
client for Unix environments. It supports conversion between different molecular
formats, substructure search, fingerprints, 2d similarity functions,
topological pharmacophore algorithms, structure modification, molecular grow,
and calculation of maximum common subgraphs.
The interaction with nails is possible via command line interface.
Nails supports the two most popular molecular formats: MDL's mol
format and Daylight's smiles. In addition, nails works with
its two internal formats: juice, and nails. The difference between these last
two formats will become clear soon.
Please refer to the special link
Fingerprint (FP) is an useful tool for fast substructure search pruning. The idea
is to calculate fingerprints for each molecule on a database, and then
compare this fingerprints to the one generated for a substructure. Because
different substructures can generate the same FP, once the algorithm tells us that
the fragment can be within the complete structure (via FP comparison), a full
substructure search is performed.
There are two special clients for fingerprint inplementation: fingernails,
which generates and reads FP, and calculates tanimoto similarities on those;
and subnails, which performs substructure search on juice's FP.
Please read more about these clients from the README file.
Let's consider we have a small set of 6 fragments that we want to search in
a database of 650 compounds. The average molecular weight of the database
is 196 g/mol. Table 2 shows the results for the substructure
search of the fragments in the database. The test was performed using a
pentium III 1 GHz machine and Linux as operating environment. Nails was
compiled using gcc 3.2 with no optimizations options. The worst-case
complexity of the substructure search algorithm is O( n^3 ).
Table 2:Substructure search results
| Fragment smile | Full search time(sec) | Fingerprint-pruned search time(sec) |
Improvement (%) |
| C1CCC2CCCCC2C1 |
5.32 |
1.81 |
66 |
| c1ccccc1 |
0.71 |
0.49 |
31 |
| OC=O |
0.36 |
0.06 |
86 |
| C1CCCCC1 |
2.09 |
1.23 |
41 |
| CCCCC1CCCNC1 |
1.77 |
0.01 |
99 |
| CC1CC(CN(C)C1)OCCC2CCCC2 |
0.74 |
0.0 |
100 |
Obviously improvement is high when the fingerprint of the fragment is not
present in the molecule fingerprint: improvement is independent from the
molecular size.
Another common application of the FPs is the calculation of 2d topological
similarity. fingernails implementation uses Tanimoto distances
on the bitsets to calculate a similarity value between two molecules.
The Tanimoto distance exhibit all required properties from distance mesures, and
its value lies within the range [1,0].
To show an example, Tanimoto distances where calculated on fingernails' FP from
a database of 7205 random pairs, and the same number of bioisosteric pairs.
Figure 1 shows the bins distribution of both, random and bioisosters pairs.
Figure 1: fingernails
similarity.
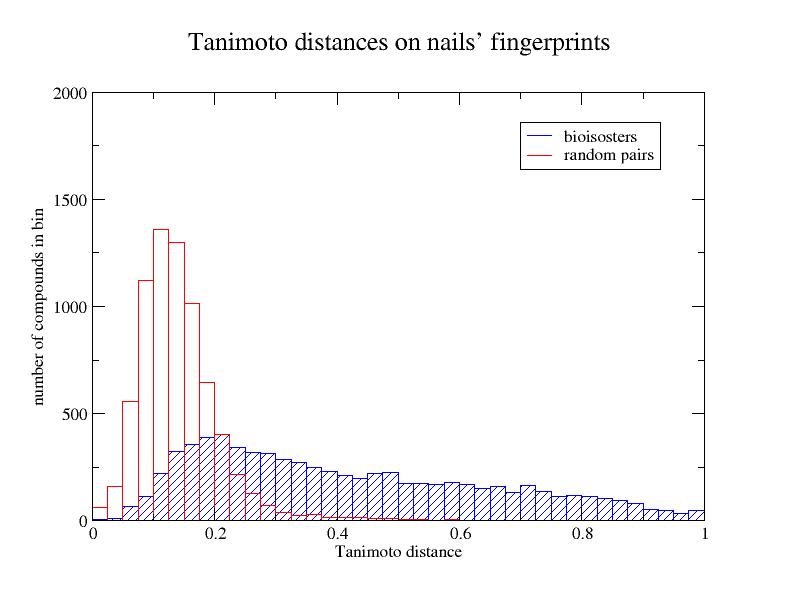 |
Figure 1 shows a clear separation of bioisosters from the random pairs. However,
there's still a big overlapping region. The abstraction achieves a statistically
satisfactory separation, with a simple calculation. Other methods are intended
to provide a better separation.
Nails implements a very simple but comprehensible description of pharmacophores,
taken from ideas expessed on [Schneider et al., 1999]. Please refer to the
paper for a throroughly description of the technique.
As a proof of concept, the algorithm was tested with the same dataset
fingernails were tested. Figure 2 shows the bins distribution of both, random and
bioisosters pairs.
Figure 2: Tanimoto distance
on the CATS vector.
 |
Figure 2 shows a similar distribution than the fingernails.
But how orthogonal both methods are? are they classifying same coumpounds to
same classes?. Figure 3 shows the Tanimoto distance on the CATS plotted against
the Tanimoto distance on the fingerprints:
Figure 3: Methods comparison.
 |
See the accumulation of of points on the right edge of the plot. Suppose we combine
both methods with a weighting function. Would be a good idea to create a function that
shift the weight to the edges of the bounds: when one of the methods is giving
a value near one of the edges, then more weight is given to that result. Have fun.
The maximum overlapping set (MOS) of a graph (or the maximum common edge subgraph),
is calculated with a stand-alone client moils, and is used to
provide 2d similarity values. Also you can print the resulting MOS(s) of two
structures. Please read the README text file or call the client with the
-h option for a synopsis on how to use it.
The algorithm was implemented with ideas given in [Raymond, 2002].
The basic idea in similarity calculations with MOS is that molecules that
share the mayority of their edges should be 2d similar.
The bad news is that the calculation of the maximum common edge subgraph is a
NP-complete problem. Some heuristics are on their way, but so far, is you're
going to calculate the MOS, be careful with the size of the molecule you're
inputing: as the molecular size grows, the problem becomes untractable.
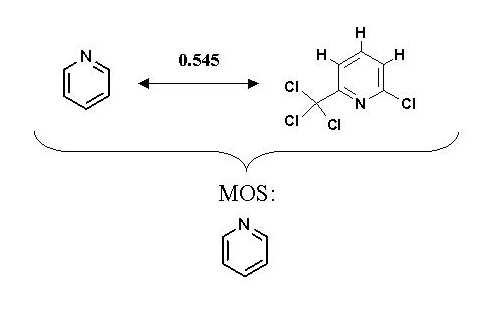
Figure 4 shows MOS distance and substructure of two molecules.
Figure 4: MOS distance and
substructure.
 |
Structure modification has its own client as well: nailgrow. Please
refer to the README file, or run it with the -h option to get a synopsis on
how to use it.
Functionalities are provided to remove fragments, add fragments, replace fragments,
etc. You can have a lot of fun with it...
Nails offers nice possibilities to work with data mining and/or classification
algorithms. In this line, we can use nails to describe molecules (whether descriptors,
pharmacophores, or 2d fingerprints) and use data-mining engines to cluster,
classify or predict biological or phisico-chemical activities.
Following are given some examples of classification algorithms:
We use the Support Vector Machines Template Library
to generate support vector machines on data obtained from the calculation of
cats pharmacophore vectors of molecules.
The idea is to calculate hyperplanes that will hopefully separate active
molecules from non-actives. Obviously the information obtained from a collection of
molecules is more general than the one obtained from just one molecule. When we make
calculations of distances from a template to a set of molecules, just the information
of one molecule is given.
The next example was performed on a database of 41 compounds (20 actives, and
21 non-actives). We use this database to generate SVMs, and later we tested
the performance classifying 43 compounds we left out for testing. Sadly we
cannot show the structures of the molecules, less the target.
Table 3:Support Vector Machines classification
| Real Cluster | Separation from the calculated hyperplane |
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
-1
|
2.16815
1.10605
1.70249
0.852959
2.29406
1.34863
0.966051
0.566114
0.666675
0.844608
0.949025
0.757625
2.41355
0.844608
-0.628612
1.2351
1.10605
2.31583
0.647329
0.289167
-0.548536
2.19804
1.81983
0.838507
-0.957572
1.17329
-0.115009
-0.52718
-2.149
-1.57006
0.183763
2.42272
-0.29059
3.35241
-0.916957
-0.117745
0.121313
-0.366695
-0.616971
-0.813379
-1.15801
-0.667753
-1.40077
|
The SVMs were able to classify correctly 34 out of the 43 compounds (79% of
the compounds).
Just your imagination is the limit for using Neural Networks in conjunction
with nails. Good examples are offered in the MolBrain
link. However, following a small example is given that show you the synergy.
Some topological descriptors of a database were calculated, and a neural network
was trained using these as the input variables. The output to fit was the
molweight of the compounds. The dataset was composed of 256 compounds, 36 of
which were randomly selected for a test set, and the rest were used to train
the NN.
Figure 5 shows the predicted values plotted against the real values of the
molweight.
Figure 5:NN prediction of molweight
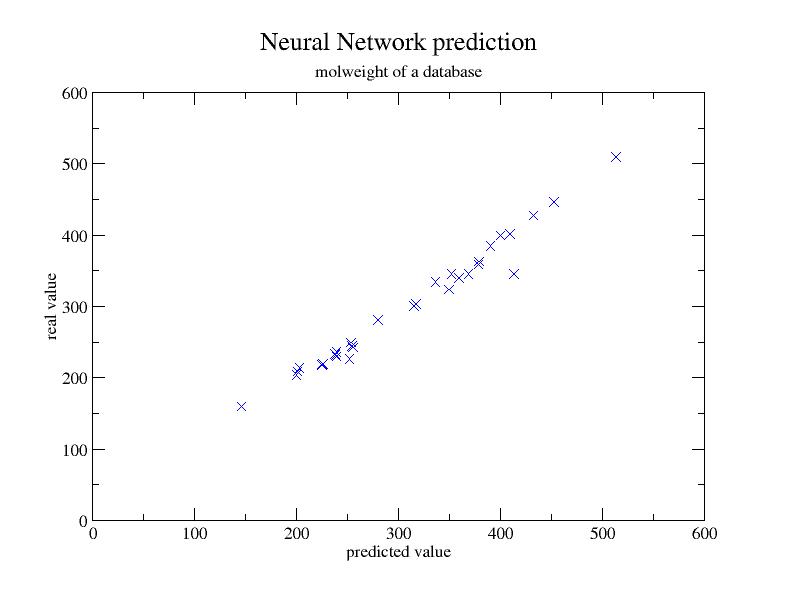 |
The correlation obtained was 0.98.
- Heuristics to calculate the MOS; then hopefully will be possible to
calculate MOS of large molecules
- Similarity calculations for 3d structures as described in [Raymond, 2003]
- Grow pharmacophore on semi-random positions given by the CATS matrix
- Schneider, G.; Neidhart, W.; Giller, T.; Schmid G.; "'Scaffold-Hopping' by Topological Pharmacophore Search:
A Contribution to Virtual Screening". Communications of Angew. Chem. Int. Ed. 1999. 38. No.19.
- Raymond, J.; Gardiner, E.; Willett, P. "Heuristics for similarity searching
of chemical graphs using a maximum common edge subgraph algorithm".
J.Chem.Inf.Compu.Sci., 2002. 42, 305-316.
- Raymond, J.; Willet, P.; "Similarity Searching in Databases of Flexible 3D
Structures Using Smoothed Bounded Distance Matrices". J.Chem.Inf.Comp.Sci.; 43.
908-916. 2003.
Copyright (c) Vladimir Josef Sykora and Morphochem AG 2003