| Reduced Molecule |
reduced_molecule
This case is a great example to show the power of the CDL. Because of CDL's generic nature,
we can create a plain molecule type, having another molecule as the type for the atoms of
this new type.
In other words, instead of writing a new class that provides this fuctionality, we just declare
a type. For example :
// First define the type of the molecular properties
typedef boost::property<mol_propsS, molecular_properties<> > mol_props_t;
// Now define the type of the atomic properties
typedef boost::property<atom_static_propsS,
::morpho::atomic_props::atomic_properties<> const *,
boost::property<indexed_mol_propS, index_mol_t> > atom_props_t;
// Note that we are using an indexed_molecule as an additional type for the atoms!
// The tag indexed_mol_propS is defined later...
// Now we just have to declare the molecule :
typedef molecule<double,mol_props_t,atom_props_t> reduced_mol_t;
#include <morpho/cdl/molecule/reduced_molecule.hpp>
 |
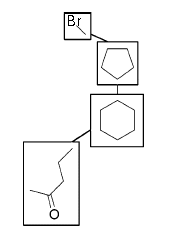 |
The function first gets all the fragments possible using the SMARTS algorithm, then it calls an user-defined functor to select which of these fragments to use to create the reduced_molecule, and then calls another user-defined functor that encodes functionality on how to use these fragments to add nodes and connetions to the reduced_molecule.
template <class Molecule, class ReducedMolecule, class ReducedMolCreator, class FragSelector> void fragmentize(const std::vector<std::string>& fragments_smart, Molecule& m, ReducedMolecule& reduced_mol, ReducedMolCreator redmol_creator, FragSelector selector);
| Parameter | Description | Models |
|---|---|---|
|
|
The SMARTS that indicates the fragments to use to subdivide the molecule | std::vector<std::string> |
|
|
The molecule to fragmentize (or subdivide) |
morpho::cdl::molecule<> |
|
|
The reduced molecule to create. This parameter should be empty. |
morpho::cdl::reduced_molecule<> |
|
|
Functor that accepts a std::vector<std::vector<size_t> > that contains the vertex indices of the fragments, and a reduced_molecule which will be created out of these fragments. Returns void. | Use make_redmol_all_nodes or make_redmol_fused_nodes |
|
|
Functor that selects the group of fragments to use to create the reduced_molecule. operator() accepts std::vector<std::vector<size_t> > which is the container that have all fragments found, and returns the same type with the selected fragments to use. |
Use largests_fragments_selector or all_fragments_selector |
int main() {
using namespace std;
using namespace morpho;
nail_juice<> j;
get_juice_from_stream(std::cin, j, 0, smiles_formatT());
// now construct the molecule
typedef molecule<> M;
M m(j);
// create the SMARTS to explode the molecule :
std::vector<std::string> fragments_smart;
fragments_smart.push_back("C1CCCCC1");
fragments_smart.push_back("C(=O)O");
fragments_smart.push_back("CNC");
fragments_smart.push_back("ONC(=O)C");
// declare the reduced molecule :
reduced_molecule reduced_mol;
// declare the functor to select the fragments :
make_redmol_fused_nodes<M,reduced_molecule> redmol_create(m);
// now call fragmentize :
fragmentize(fragments_smart,m,reduced_mol,redmol_create,all_fragments_selector());
// Now we have fragmentized molecule 'm' in a reduced_molecule 'reduced_mol'
// To see its content, we can :
reduced_mol_t::vertex_iterator vi, vi_end;
tie(vi,vi_end) = reduced_mol.vertices();
while(vi!=vi_end) {
std::cout << "--- New atom ---. Index : " << *vi << std::endl;
std::cout << write_nail(
AtomProperty<index_molS,reduced_mol_t::atom_type>::get(reduced_mol.get_atom(*vi))
,smiles_formatT()) << std::endl;
std::cout << "-- Indexed molecule, dissociations :\n";
const AtomProperty<index_molS,reduced_mol_t::atom_type>::result_type::dissociations_t&
diss = AtomProperty<index_molS,reduced_mol_t::atom_type>::get(
reduced_mol.get_atom(*vi)
).get_dissociations();
for(int i=0;i<diss.size();++i) {
std::cout << "--> dissociation : " << diss[i] << std::endl;
}
std::cerr << "--- Adjacent vertices : ---\n";
reduced_mol_t::adjacency_iterator ai, ai_end;
tie(ai,ai_end) = reduced_mol.adjacent_vertices(*vi);
while(ai!=ai_end) {
std::cerr << *ai << ",";
++ai;
}
std::cout << std::endl << "--end adjacency\n";
++vi;
}
return 0;
}
... y voile
 |
 |
 |